来源:Cytiva
作者: Mats Lundgren, Cytiva
如何驾驭生物制药领域的发展带来的工艺开发变化
生物制药行业发展迅猛,特别是在生产方面。单克隆抗体 (mAb) 或由克隆自单细胞的相同免疫细胞制造的抗体对开发人员来说已不再是挑战,mAb 已成为行业标准,具有成熟的研究、开发和生产工艺,实践证明非常可靠,具有可放大性。在当今生物制药领域,开发人员经常遇到各种各样、结构复杂的生物分子。各种新型生物制品在进入药物管线的同时,也对上游、下游和分析开发领域的现有生产工艺构成了挑战。
了解这些不同分子会带来哪些挑战以及如何应对这些挑战,是确保它们安全高效地用于患者的关键。
不断增长的分子多样性
在大多数制药公司中,生物制品目前占据药物开发管线的很大比例。尽管生物制品不是新的药物类型,但最近的技术进步与人体生物学知识的发展推动了这一领域的发展,令其受到关注。专家认为,由于新型生物制品结构复杂,具有靶点结合和疗效特异性,因此在解决疑难医疗需求方面极具潜力。
生物药剂通常定义为利用活生物体生产的药物或含有活生物体组分的药物。除 mAb 外,还有许多其他种类的生物药物,包括替代抗体类型(抗体片段、抗体-药物偶联物、双特异性抗体)、重组蛋白(例如胰岛素和人体生长激素等激素)、细胞疗法(例如 CAR-T 细胞、干细胞疗法)和病毒载体(某些疫苗和基因疗法)。
下一代抗体衍生分子和病毒载体克服了简单小分子药物和 mAb 的一些治疗局限性,但同时也对工艺开发带来了一定的困难。
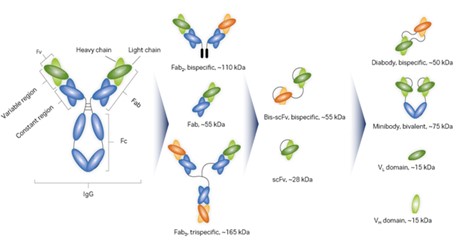
图 1. 图中所示为抗体衍生生物药剂的分子多样性示例。
开发可放大性工艺所面临的挑战
这些分子中有许多来源于学术领域,依靠创新为药物开发带来了突破性的发现。然而,学术实验室通常不是为优化工艺开发或提高生产效率而设立的。此外,基础研究的开展并非总是会考虑监管机关能否接受。
因此,能否将突破性的发现转化为可放大性工艺,已成为新型疗法生物制造商面临的首要挑战。Mats Lundgren 博士表示,“在我看来,作为开发人员,我们面临的最大挑战之一是放大新分子的生产规模,并确保生产工艺可为临床试验和商用方案提供足够高的优质原料药收率”,Mats Lundgren 博士是 Cytiva 的客户应用主管,在疫苗和病毒载体处理方面有超过 20 年的经验。
此外,一些早期的生产工艺都是基于旧的技术。这可能导致工艺不易放大或不经济。例如,使用胎牛血清作为病毒载体的细胞培养添加剂在学术实验室中可能司空见惯,但这种做法在生物制造中却行不通,因为生物制造行业中关于污染和患者安全性的严格法规禁止使用动物源性组分。另一个示例是使用离心法或尺寸排阻层析法进行纯化。虽然这些技术适用于小规模处理,但不适用于大规模生产。Lundgren 表示,这一切让我们认识到,应该从一开始就采用适合放大的技术 – 对此我们将在后面更详细的介绍。
质粒 DNA 的生产是困扰病毒载体生产的另一个瓶颈问题。病毒载体是通过 DNA 重组技术开发的病毒,其作用是将遗传物质递送至靶细胞。病毒载体通常用含有目标基因的质粒 DNA(一种与细胞染色体 DNA 不同且可独立复制的小型环状双链 DNA 分子)进行细胞转染的方式生产。由于质粒 DNA 是关键原料,生产的基于病毒载体的治疗药物越多,对质粒 DNA 稳定供应的需求就越大。但问题在于,质粒 DNA 必须在符合《药品生产质量管理规范》(GMP) 的工厂或至少在高质量质粒 DNA 生产区域中生产和纯化。因此,此类生产的放大会给药物开发企业带来很大压力,他们需要提高生产能力以满足对高质量质粒 DNA 不断增长的需求。
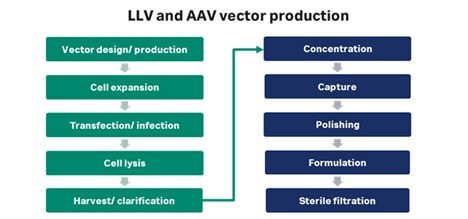
图 2. 慢病毒 (LV) 和腺相关病毒 (AAV) 病毒载体的制备方法示意图。
时间窗更短,缺少平台,面临纯化挑战
随着开发的时间线变得越来越短,工艺开发的效率已成为一个必要的成功条件。Lundgren 指出,“mAb 技术刚起步时,工艺开发需要数年的时间”。“但现在必须在极短的时间窗内完成开发,而需要处理的分子却困难得多。”
此外,新分子没有开发路线图;与旧的 mAb 开发方式一样,目前还没有合适的平台技术。Lundgren 指的是基于病毒载体的新型疫苗这一近几年才开辟的全新领域。研发人员需要花费大量的时间和精力来寻找最佳生产方法以及探索如何应用这些方法。即使在病毒载体(腺病毒、腺相关病毒、慢病毒、逆转录病毒等)这样的微小领域内,分子种类繁多也会使问题更加复杂。
另一方面,由于有些新分子形式的存在引入了新的杂质,需要减少此类杂质的含量,因而药物质量要求也在增加。特别是当某种分子可能存在多种变体时,必须证明这些变体均不会影响治疗的安全性和有效性。如果有影响,需要制定质量控制措施来管理/减少这些变体。
例如,以双特异性抗体为例,在表达过程中可能会形成抗体的多种变体。开发人员最终可能会得到重链和轻链的同源二聚体而非异源二聚体,或轻链与重链的随机结合。导致与 mAb 生产相比收率降低。美国 Regeneron 副总监 Andrew Tustian 博士认为,纯化双特异性抗体的主要痛点之一是杂质含量增加。他表示,杂质通常与目标分子非常相似,所以难以分离和去除。5
克服新生物分子工艺开发挑战的建议
Lundgren 认为,克服工艺开发挑战的第一步也是最重要的一步是进行彻底和仔细的可行性研究。这将涉及计算应生产多少物料以及应使用哪些技术才能实现必要的结果。他表示,“您需要非常了解您的市场和技术,并且要做好功课,这样才能在预算范围内生产出成功的产品”。
接下来是使用相关的工艺开发工具找到最佳工艺和方案,例如使用质量源于设计 (QbD) 方法。在此方面,Lundgren 认为可以从多年来单克隆抗体领域的发展历程中学到很多东西。例如使用符合法规要求的细胞培养组分,应使用化学成分明确的细胞培养基,而不是动物源性血清。化学成分明确的培养基需要确定所有组分,并知道它们的确切浓度。使用化学成分明确的培养基是一种“良好细胞培养规范”(GCCP),可以帮助制定体外方法的通用标准。
mAb 领域已非常成熟,即使在研发过程中也不允许使用动物源性组分。但学术界在研究新生物分子时则不会遵循这样的要求。学术研究人员并不总是熟悉法规约束,他们倾向于使用多年形成的传统方法。Lundgren 介绍说,有人认为化学成分明确的细胞培养基获得的收率较低,现在看来这种看法是错误的。他表示,“我们有非常好的不含动物源性组分的纯细胞培养基,而且它们照样非常好用”。与该领域的专业供应商合作有助于避免原料不合适带来的风险,而且可以获得高滴度。
如果在设计之初就采用可放大性技术(如层析法),可在很大程度上克服新分子的工艺开发挑战。例如,使用多模式层析填料,如 Cytiva 的 Capto™ Core 700,可以帮助提高速度和改善病毒载体的工艺经济性。再例如,可以使用一次性技术和设备,以减少清洁要求并改善批次周转时间。8为扩大质粒 DNA 的生产规模,Cobra 和 Cytiva 最近合作开发了“Capto PlasmidSelect”层析填料,这种填料能提高可放大性和效率,同时将产品成本保持在可接受的水平。4
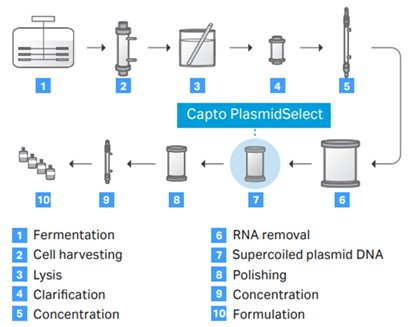
图 3. 从发酵到制剂的高质量超螺旋质粒 DNA 生产工艺示意图。
产品分析和表征的重要性
由于复杂生物制品尤其需要高质量的精密分析,而且高质量的分析极为关键,因此在工艺开发过程中必须使用优良的分析工具。Lundgren 强调,“没有优良的分析测量就无从谈起,更谈不上优化工艺”。“虽然具体使用的工具与分子有关,但拥有适用所分析分子的不同分析工具的可靠组合非常重要。”
病毒载体需要用到许多分析方法进行不同方面的分析,例如鉴别、效价、纯度、安全性和稳定性。物理性病毒滴度(给定体积液体中病毒的量)可通过 DNA 杂交、实时 PCR (qPCR、ddPCR)、SPR 技术(如 Biacore™ 或高效液相层析法 (HPLC))计算,而功能性病毒滴度可通过噬菌斑形成或荧光焦点试验测量。杂质可通过质谱法或电子显微镜检查法来检测,而安全性可以通过 Southern 印迹、qPCR 或体外/体内试验来检查。确定载体稳定性时可采用显微镜检查、电位测定法和渗透压测定法等技术。9
显然,检测和表征生物药剂及其杂质(产品与工艺相关杂质)需要用到各种分析技术。对于大多数新分子和复杂分子而言,必须开发定制化测定技术,同时考虑测定速度和稳健性。
结论
实施可放大性技术并遵循 QbD 原则是应对生物药剂开发挑战的有力措施。为避免后期问题和产生不必要的成本,有必要投入足够的时间和资源开放适当的可放大性工艺。Lundgren 建议开发人员尽早对工艺经济性进行建模,“在开发项目和商业概念时,尽早进行这些模拟非常重要。据我们所知有许多公司不会进行模拟,不幸的是,这些公司最终都无法获得经济可行的分子生产技术”。
扩展阅读
参考文献
1. Jalal, B. Realizing the Promise of Biologics. Harvard Health Policy Review, 2017.
2. Oo, C., Kalbag, S.S. Leveraging the attributes of biologics and small molecules, and releasing the bottlenecks: a new wave of revolution in drug development. Expert Review of Clinical Pharmacology, 2016, 9:6, 747-749.
3. Biological Therapies for Cancer. National Cancer Institute, 2018.
4. Charlesworth, J., Hitchcock, T., Tharia, H. et al. Purifying plasmid DNA using a modern chromatography resin. Cytiva Bioprocessing Knowledge Center.
5. Westman, D. Bispecific antibody purification: insights and case studies. Cytiva Bioprocessing Knowledge Center, 2019.
6. Tustian, A.D., Endicott, C., Adams B. et al. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs, 2016, 8(4): 828-838.
7. van der Valk, J., Brunner, D., De Smet, K. et al. Optimization of chemically defined cell culture media – Replacing fetal bovine serum in mammalian in vitro methods. Toxicology in Vitro, 2010, 24, 1053-1063.
8. Darby, N., Schmidt, S.R., Lidén, P. et al. Preparing for the future–visions and insights for biomanufacturing. Cytiva Bioprocessing Knowledge Center.
9. King, D., Schwartz, C., Pincus, S. et al. Viral Vector Characterization: A Look at Analytical Tools. Cell Culture Dish, 2018.
10. Kramberger, P., L. Urbas, Strancar, A. Downstream processing and chromatography based analytical methods for production of vaccines, gene therapy vectors, and bacteriophages. Human Vaccines & Immunotherapeutics, 2015, 11(4): 1010-21.